
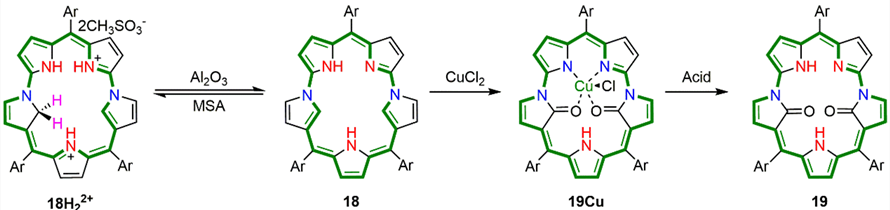

扩环卟啉为探索包括莫比乌斯芳香性、反芳香性体系以及稳定自由基在内的非经典π电子结构提供了多功能骨架。除了这些静态电子态,近期的关注已转向扩环卟啉骨架结构的蜕变,即一种大环骨架转变为另一种,从而实现结构和功能的不连续变化。
尽管已有热触发和金属触发骨架重排的显著例子,但这些过程仍需依靠键的断裂或重排。相比之下,通过可控氧化进行的骨架编辑提供了一种截然不同的策略:它并非破坏大环,而是通过原子的引入实现共轭路径的改变(图1)。然而,这类转化发展尚不充分,尤其对于反芳香性扩环卟啉而言,其高反应活性和多重竞争的共轭路径使得选择性修饰变得复杂。在此背景下,N错位扩环卟啉提供了独特的骨架,因为氮错位吡咯单元引入了具有电子偏倚的内α位,使其易倾向于发生选择性官能化。
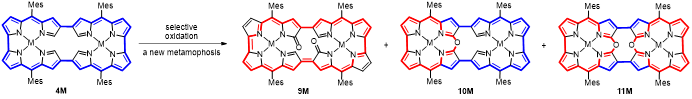
图1:四重N错位八元卟啉的氧化蜕变
基于上述分析,课题组通过对四重N错位八元卟啉内α位的氧化修饰,突破了需要键的断裂或重排才引起大环骨架改变的壁垒,将氧化骨架编辑确立为一种新的诱导扩环卟啉蜕变的通用策略,实现了大环共轭电子体系的可控重排。相关成果近日在国际化学顶级期刊Angew. Chem. Int. Ed.上发表,题为“Oxidative Porphyrinoid Metamorphosis of Fourfold N-Confused [32]Octaphyrin Bis-Metal Complex to Doubly Linked 10-Oxacorrole Metal Complex Dimer”。
首先,通过Yamamoto偶联反应合成了基于Bisdipyrrin模块桥联的二聚体、三聚体和四聚体(图2)。二聚体4Ni和4Pd以经典的32π反芳香性体系为主。X射线单晶衍射表明,二聚体4Pd的骨架结构呈现出平面矩形构象。与之相反,三聚体5Pd呈现显著扭曲的三角形几何构型,而四聚体6Pd则采取螺旋扭曲的“8”字拓扑结构(图3)。实验结果表明,尽管5Pd和6Pd其形式上具有48π和64π电子共轭路径,但由于其扭曲的空间结构破坏了它们的有效共轭路径,使分子呈现出非芳香性。
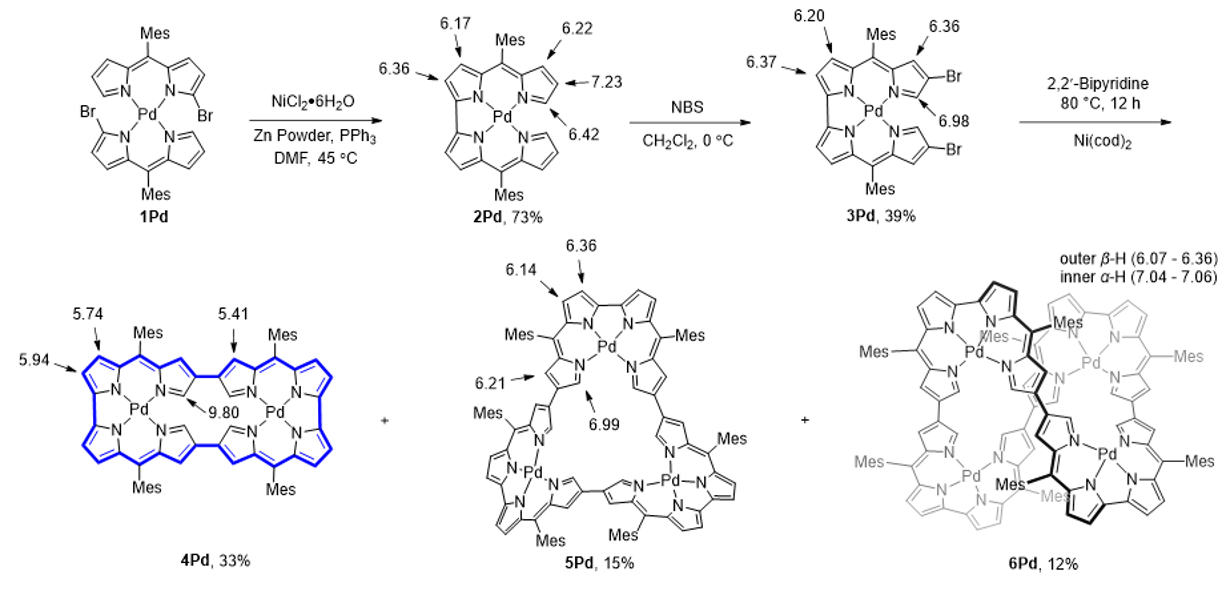
图2:化合物4Pd、5Pd和6Pd的合成
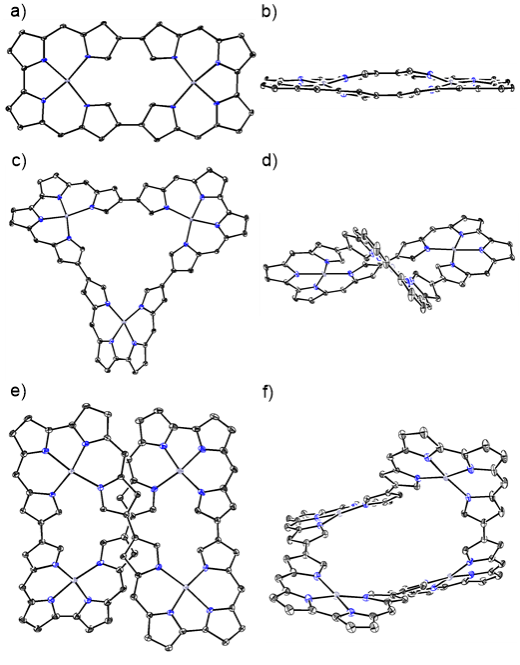
图3:化合物4Pd、5Pd和6Pd的单晶结构
在前期研究的基础上,作者进一步探索了4Pd的氧化转化,重点针对N错位吡咯的内α位进行官能化。在CH2Cl2/MeOH中用Cu(OAc)2处理4Pd,得到了甲氧基化衍生物7Pd和8Pd,同时伴有少量二酮产物9Pd的生成(图4)。X射线晶体学分析证实,甲氧基被引入到内α位,整体大环骨架得以保持(图5)。同时,7Pd和8Pd均保留了整体的32π电子环路,呈现出反芳香性的特质。相反,二酮衍生物9Pd则具有明显的芳香性。
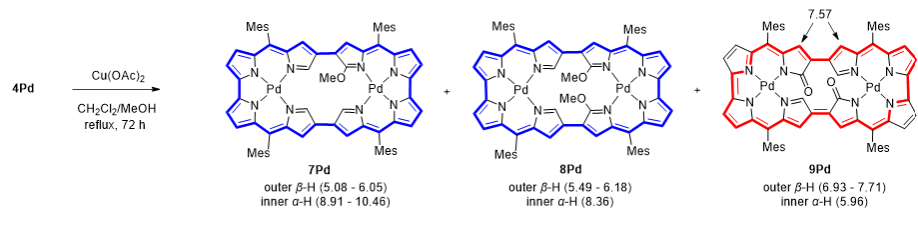
图4:化合物7Pd、8Pd和9Pd的合成
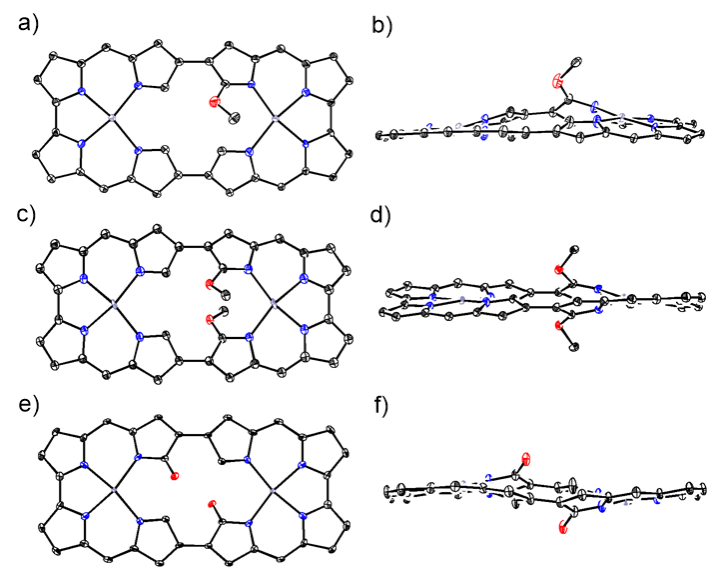
图5:化合物7Pd、8Pd和9Pd的单晶结构
在低温下用间氯过氧苯甲酸(m-CPBA)氧化4Pd时,观察到了更为引人注目的转化(图6)。X射线晶体学分析揭示,10Pd的大环内嵌有一个单一的10-氧杂咔咯亚单元,而11Pd则是一个由10-氧杂咔咯亚单元双重连接形成的二聚体(图7)。这些化合物展现出独特的电子结构,其特征是分子局部共轭路径与分子整体共轭路径的共存。10-氧杂咔咯亚单元具有18π芳香性回路,而分子整体骨架则保留32π反芳香性回路。
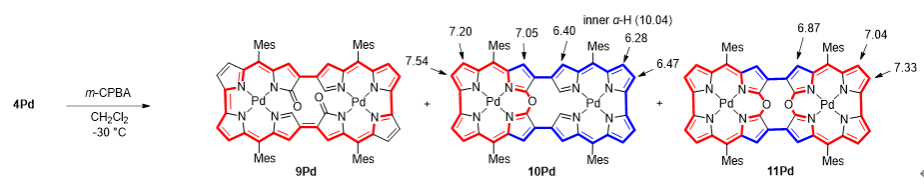
图6:化合物9Pd,10Pd和11Pd的合成
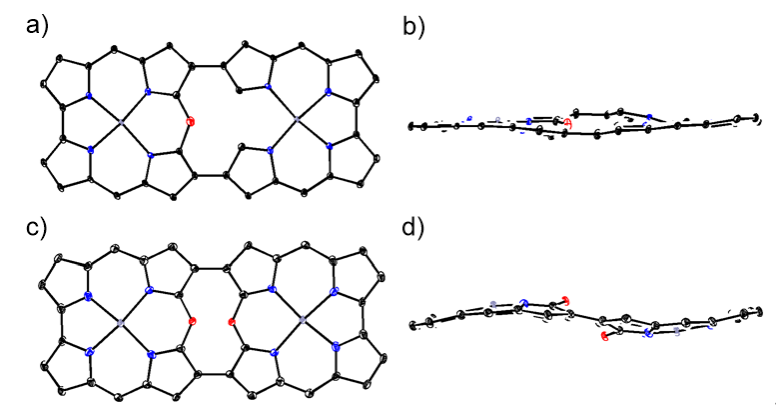
图7:化合物9Pd,10Pd和11Pd的单晶结构
利用甲基三氧化铼(Me3ReO3)与过氧化氢(H2O2)的组合实现了4Ni的高效转化(图8),生成了二酮化合物9Ni、单-10-氧杂咔咯10Ni、双-10-氧杂咔咯11Ni以及二酮-单-10-氧杂咔咯12Ni。将11Ni置于相同反应条件下处理,以25%的产率得到12Ni,表明12Ni是11Ni的次级产物。结构表征确认,10Ni和11Ni与其钯类似物高度相似,说明这一蜕变过程在不同金属中心上具有普适性。值得注意的是,化合物12Ni表现出增强的抗磁环电流,这源自分子局部18π芳香性10-氧杂咔咯单元与分子整体30π芳香性回路的组合。这导致其1H NMR谱中出现显著的低场位移,凸显了多重共轭路径的累积效应。
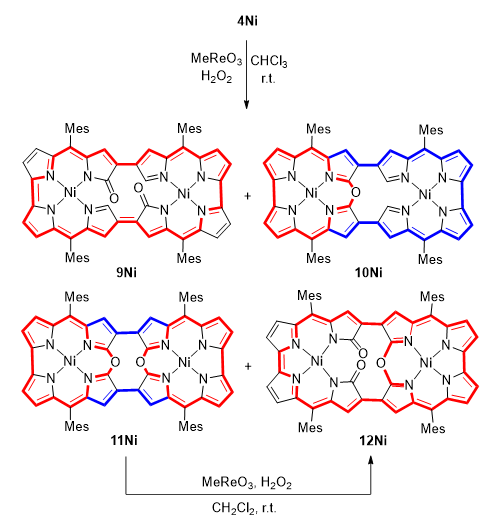
图8:化合物9Ni,10Ni,11Ni和12Ni的合成
总之,通过氧化将反芳香性扩环卟啉4M转化为结构和电子性质截然不同的扩环卟啉这一骨架编辑策略,代表了一种此前未被认识的扩环卟啉转变形式:反芳香性八元卟啉骨架在此过程中转变为芳香性二酮八元卟啉及嵌入10-氧杂咔咯的八元卟啉。该过程由N错位吡咯内α位的选择性氧化所触发,导致氧原子插入并随后引发π共轭体系的重组。重要的是,这一转化伴随着电子结构的根本性变化,即从分子整体反芳香性转变为分子整体芳香性(30π)体系,或转变为局部芳香性与整体反芳香性共存的体系。这些发现表明,氧化骨架编辑为在扩环卟啉体系中同时调控结构和芳香性提供了一种强大且通用的方法。
论文第一作者为硕士研究生刘云龙,通讯作者为Atsuhiro Osuka教授和宋建新教授。
论文链接: https://onlinelibrary.wiley.com/doi/10.1002/anie.4104555